This protocol is deprecated please see latest V5 protocol.¶
Protocol for Making ChronoSeq V4 Beads by Modifying Dropseq beads for Bulk Time Series.¶
This protocol is for making 400μl of Beads for each Time-Tag. Ideal for Testing in Bulk Time Series.
Bead Modification Workflow |
|---|
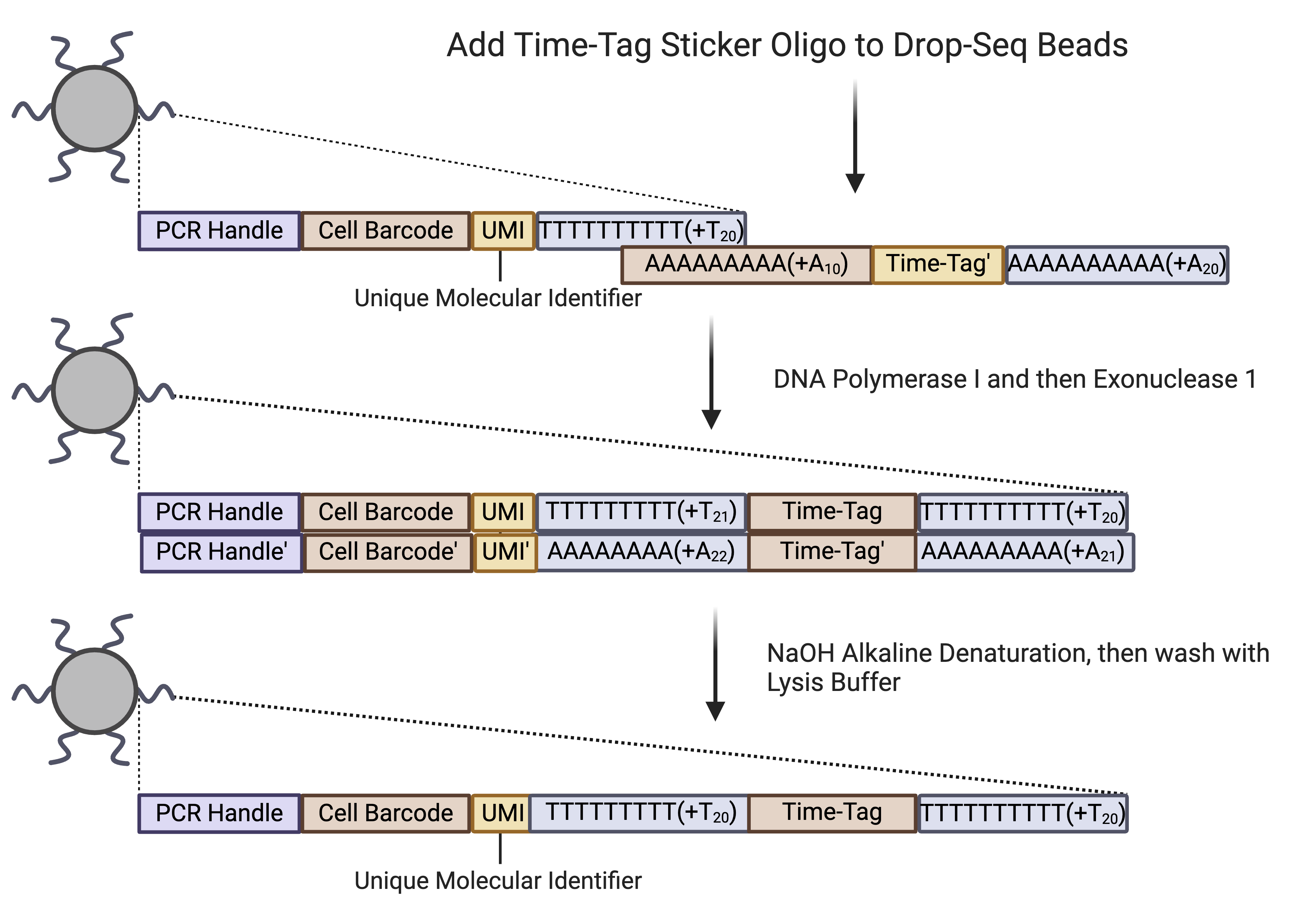 |
Sequences are added by using an oligo that binds to the existing PolyT region on Drop-seq beads. The 3’ end of the Drop-seq beads are then extended to the reverse compliment of the oligo using E. Coli DNA Polymerase I. Single-stranded DNA are removed using Exonuclease 1. This leaves only double-stranded DNA on the beads that cannot capture RNA. Therefore, Alkaline denaturation is used to make the double stranded DNA single-stranded. These beads are then resuspended in lysis buffer and can be used directly with our device.
Dropseq beads Properties and Sticker Sequence¶
5’–Bead–Linker-‐TTTTTTTAAGCAGTGGTATCAACGCAGAGTAC JJJJJJJJJJJJNNNNNNNN TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-‐3’ Beads were ordered from Chemgenes and synthesized at a 10μmol Starting Scale.
- We ordered 3 Units of Dropseq beads for making these beads.
- Catalog Number: Macosko-2011-10(V+)
- Each unit gives us between 12-15ml of Beads @450beads/μl
- We used 400μl of beads at @450beads/μl per Time-Tag
Update: New Supplier¶
- LGC Biosearch Technologies have started offering these beads.
- If you use the LGC beads make sure you use the same amount of beads for each reaction.
- These beads have a longer UMI length of 14 instead of 8 for Chemgenes. The sequence for these beads is as follows:
- 5’–Bead–Linker-‐TTTTTTTAAGCAGTGGTATCAACGCAGAGTAC JJJJJJJJJJJJNNNNNNNNNNNNNN TTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-‐3’
- They also claim better quality control and fewer particles that can cause a blockage in the Microfluidic Chip.
- You can order them here.
Time-Tagged Sticker Oligos for making ChronoSeq V4 Beads using Dropseq beads¶
All Oligos were ordered from IDT
- 25nmol Scale
- Standard Desalting
- SEQ1_TTGG: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA TNTHGHGB AAAAAAAAAAAAAAAAAAA
- SEQ2_CCTT: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA CDCDTNTB AAAAAAAAAAAAAAAAAAA
- SEQ3_GGAA: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA GHGHANAB AAAAAAAAAAAAAAAAAAA
- SEQ4_TTCC: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA TNTDCDCB AAAAAAAAAAAAAAAAAAA
- SEQ5_TTAA: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA TNTSANAB AAAAAAAAAAAAAAAAAAA
- SEQ6_TTTT: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA TVTVTVTB AAAAAAAAAAAAAAAAAAA
- SEQ7_CCAA: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA CDCKANAB AAAAAAAAAAAAAAAAAAA
- SEQ8_CCGG: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA CDCWGHGB AAAAAAAAAAAAAAAAAAA
- SEQ9_CCCC: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA CDCDCDCB AAAAAAAAAAAAAAAAAAA
- SEQ10_GGTT: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA GHGMTNTB AAAAAAAAAAAAAAAAAAA
- SEQ11_GGCC: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA GHGWCDCB AAAAAAAAAAAAAAAAAAA
- SEQ12_GGGG: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAA GHGHGHGB AAAAAAAAAAAAAAAAAAA
Solutions and Buffers to Make¶
Use Distilled Water for all these Buffers. Please also filter using a 40μm Cell Strainer.
STOP Buffer:
- 10 mM Tris HCl pH 8.0 + 1 mM EDTA
- 0.5% Sarcosyl
- 330mM KCl
WASH Buffer:
- 10 mM Tris HCl pH 8.0 + 1 mM EDTA
- 0.01% Tween-20
- 330mM KCl
TE-SDS:
- 10 mM Tris HCl pH 8.0 + 1 mM EDTA
- 0.5% SDS
TE-TW:
- 10 mM Tris HCl pH 8.0 + 1 mM EDTA
- 0.01% Tween-20
IDTE Buffer:
- 10 mM Tris HCl pH 8.0 + 0.1mM EDTA
Buffer for Dissolving and Diluting Oligos Ordered from IDT
- 1M DTT:
- Dissolve 1.55g solid DTT in 10ml of Deionized Water. Hazardous Chemical Read SDS. Work in Fume Hood
More economical to make yourself from Solid DTT. Make 50ml or more at a time and store in 10ml Aliquots for upto an year. DTT is not stable at room temperature. Make this solution quickly and store at -20°C, unless you plan to use it immediately to make another Buffer.
- 10X NEB Buffer 2:
- 500 mM NaCl
- 100 mM Tris-HCl pH 7.9
- 100 mM MgCl2
- 10 mM DTT
Make in Bulk for Scaling up reaction. It is more economical than buying NEB Buffer 2. Make 100ml or more at a time and store in 11ml Aliquots for upto an year.
DTT is not stable at room temperature. Make this solution quickly and store at -20°C.
Best Practices:¶
- Remember to put back the reagents back to their storage location at 4°C or -20°C once you are done with using them.
- Get fresh new microtips/sterile-disposable pipettes after each step, unless mentioned otherwise.
- Important to avoid cross-contamination.
Equipment Used:¶
- Our lab uses the refrigerated Sorval ST8R Centrifuge. You can find CAD files to print some of the parts here.
- We bought the 50ml Inserts and Printed the Rest.
- You can modify the CAD files to print the 50ml Inserts as well.
- Use PLA or TPU for the prints.
- Microcentrifuge we used was the Fisherbrand™ accuSpin™ Micro 17/17R Microcentrifuge
- Labnet Rotating Mixer
- CELLTREAT Pipette Controller
- Labnet Vortex
- Eppendorf Pipettes 6-Pack
- VWR Scientific Products 1545 General Purpose Incubator (Discontinued Product)
- Pipette Tips:
- Larger Volume Pipettes:
- Disposable Sterile Falcon Tubes:
- Disposable Sterile 250ml GL45 Bottles, Individually Wrapped
Protocol for Bead Modification¶
Preparation of Beads¶
- Use the instructions in this section to make 10μM stocks for the Sticker Oligos ordered from IDT, using the IDTE buffer.
- Follow the ChronoSeq beads preparation protocol and suspend the Dropseq Beads from the Alcohol stock at -20°C to 1X Lysis buffer at 4°C.
- You will need about 400μl of beads @450beads/μl per Time-Tag for this protocol.
- Spin Down Dropseq beads with 450beads/μl suspended in 1X Lysis buffer.
- Turn on the UV and airflow for the Cell Culture Hood. Leave for 15 minutes.
- Inside the Cell Culture Hood: Shake Each Tube to make sure the beads are evenly suspended . For each Tube using a Sterile/Particle Free Rainin P1000 pipette tip transfer 400μl of beads to a Sterile 1.5ml Eppendorf Tube.
- Put the main 50ml Tubes with Dropseq beads back in their storage location at 4°C once you are done.
- Turn off the Cell Culture Hood and move to your bench. Take the eppies with 400μl beads at 450beads/μl with you.
This is a STOPPING STEP. You can store the Beads in 1X Lysis Buffer at 4°C and continue later.
DNA Polymerase I Isothermal Extension¶
- Get the eppies with 400μl of Dropseq beads suspended in 1X Lysis buffer.
- Set an incubator to 34°C
- Get the dNTP, 10μM Sticker Oligos and NEB Buffer 2 from -20°C and leave on your bench to melt. Put on ice once melted.
💡 Tip: Make sure you have enough volume of dNTPs, Oligos and Buffer for your experiments.
- Get the DNA Polymerase I from -20°C and put it on ice.
- Make 1.25X NEB Buffer 2 and put on ice.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Remove 300μl of Lysis buffer without disturbing the beads.
- Now wash the beads 3 Times with 1ml 1.25X NEB Buffer 2:
- Add 1ml 1.25X NEB Buffer 2. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml 1.25X NEB Buffer 2. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml 1.25X NEB Buffer 2. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Without disturbing the beads remove 400μl of Buffer.
- Estimate the Volume using the P1000.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Remove liquid without distrubing the beads to make the final Volume 320μl.
- Now add:
- 20μl of 10μM Sticker Oligo for ChronoSeq-V4. Pipette up and down several times to mix well.
- 20μl dNTP mix from NEB
- 39μl Distilled Water
- 1μl DNA Polymerase I . Use the P2.5 , Pipette up and down several times to mix well.
- Vortex the Beads at full speed for 2 seconds to mix everything well.
- Now Incubate at 34°C with Rotation for 1.5 hours.
- Set the incubator to 37°C after you are done using it.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Without disturbing the beads remove 200μl of Buffer.
- Wash the beads once with STOP Buffer and then twice with WASH Buffer:
- Add 1 ml of STOP Buffer to the eppie.
- Flip the tube upside down a few times and then Vortex the beads at full speed for 2 seconds.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of WASH Buffer. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of WASH Buffer. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
This is a STOPPING STEP. You can store the Beads in WASH Buffer at 4°C and continue later.
Exonuclease 1 Digestion¶
- Make sure the incubator is Set to 37°C.
- Get the Exonuclease 1 Buffer -20°C and leave on your bench to melt. Put on ice once melted.
💡 Tip: Make sure you have enough volume of Exonuclease 1 Buffer for your experiment.
- Get the Exonuclease 1 from -20°C and put it on ice.
- Make 1.25X Exonuclease 1 Buffer and put it on ice.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Wash twice with 1ml of 1.25X Exonuclease 1 Buffer:
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1 ml of 1.25X Exo 1 buffer to the eppie. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1 ml of 1.25X Exo 1 buffer to the eppie. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Remove 400μl of Liquid from the eppie without disturbing the beads.
- Without touching the beads, slightly pipette up and down using the P1000 to resuspend the beads in solution.
- Now estimate the Volume in the eppie by using the P1000.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Using the estimated volume in the eppie, make the final volume of the eppie 320μl without disturbing the beads.
- Then add the following to the eppie :
- 30μl Distilled Water
- 50μl Exonuclease 1 . Pipette up and down many times to mix well.
- Vortex the Beads to Mix everything Well.
- Now Incubate at 37°C with Rotation for 2 hours.
- Set the incubator to 42°C after you are done using it.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Without disturbing the beads remove 200μl of Buffer.
- Wash the beads once with TE-SDS and then twice with TE-TW:
- Add 1 ml of TE-SDS to the eppie.
- Flip the tube upside down a few times and then Vortex the beads at full speed for 2 seconds.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of TE-TW. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of TE-TW. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
This is a STOPPING STEP. You can store the Beads in TE-TW at 4°C and continue later.
- Wash the beads twice with Distilled Water:
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of Distilled Water. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
- Add 1ml of Distilled Water. Vortex the beads at full speed for 2 seconds to resuspend the beads if necessary.
- Spin down the beads using a tabletop centrifuge @ 2500xg for 1 minute.
- Carefully remove 1ml of Liquid without disturbing the beads.
Protocol for Converting dsDNA on the Beads to ssDNA using Alkaline Denaturation¶
- Sodium Hydroxide is a very dangerous Chemical it can melt your Skin and turn your body into Soap. You need to be extra careful.
- Wear Lab Coat, Safety Glasses, Face-Sheild, Double Nitrile Gloves and work in the Fume Hood.
- Tape the First Layer of Gloves to your labcoat to avoid exposing your skin accidentally.
- Execute the cell below to watch the video.
- Put on a second layer of gloves on top after you have taped the first layer.